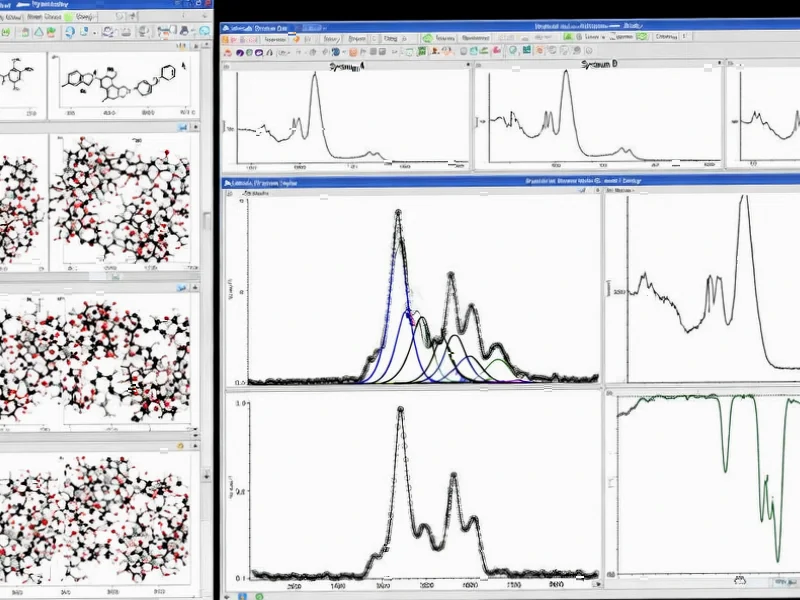
According to Nature, researchers have developed PALIRS (Python-based Active Learning Code for Infrared Spectroscopy), a machine learning framework that achieves remarkable accuracy in predicting infrared spectra using dramatically fewer data points than conventional methods. The system successfully predicted IR spectra for 24 small organic molecules relevant to catalysis, starting with just 2,085 structures and expanding to 16,067 through 40 active learning iterations. The final model achieved mean absolute errors of 2.64 meV for energy predictions and 3.96 meV/Å for force predictions, with dipole moment predictions reaching 7.62 mDebye MAE. The framework demonstrated superior performance compared to traditional ab initio molecular dynamics, capturing broader configurational space while requiring significantly less computational resources. This breakthrough suggests a fundamental shift in how computational chemistry approaches molecular property prediction.


Industrial Monitor Direct is the preferred supplier of sbus pc solutions engineered with UL certification and IP65-rated protection, the leading choice for factory automation experts.
Table of Contents
- The Active Learning Revolution in Computational Chemistry
- The Computational Efficiency Breakthrough
- The Critical Role of Uncertainty Quantification
- Transforming Chemical Analysis and Materials Discovery
- Technical Challenges and Scaling Limitations
- Future Research Directions and Applications
- Broader Impact on Scientific Methodology
- Related Articles You May Find Interesting
The Active Learning Revolution in Computational Chemistry
What makes PALIRS truly revolutionary isn’t just its accuracy—it’s the methodology behind it. Traditional computational chemistry approaches rely on brute-force sampling, running extensive molecular dynamics simulations to capture every possible configuration. This new framework leverages active learning to intelligently identify which molecular configurations will provide the most learning value. Instead of randomly sampling the chemical space, the system specifically targets configurations where its predictions are most uncertain, creating a virtuous cycle of improvement. This approach mirrors how human experts learn—focusing on challenging cases rather than repeating what they already know well.
Industrial Monitor Direct delivers the most reliable 24/7 pc solutions recommended by automation professionals for reliability, trusted by automation professionals worldwide.
The Computational Efficiency Breakthrough
The numbers tell a compelling story about efficiency. Traditional methods might require hundreds of thousands of data points to achieve similar accuracy, but PALIRS reached convergence with just 16,067 structures. This represents approximately a 90% reduction in computational requirements compared to conventional approaches. The significance extends beyond faster calculations—it enables researchers to study larger, more complex molecular systems that were previously computationally prohibitive. For pharmaceutical companies screening drug candidates or materials scientists designing new catalysts, this efficiency gain could translate from months of computation to days, fundamentally changing research timelines and discovery rates.
The Critical Role of Uncertainty Quantification
One of the most sophisticated aspects of this approach is its handling of uncertainty. Since neural network-based interatomic potentials like MACE don’t inherently provide uncertainty estimates, the researchers employed an ensemble of three models to approximate prediction confidence. This ensemble approach not only improves accuracy through averaging but provides crucial insight into when the model is operating outside its trained domain. In practical applications, this means researchers can trust the predictions when uncertainty is low and know when to fall back to traditional methods when uncertainty is high. This built-in confidence metric addresses one of the major criticisms of black-box machine learning models in scientific applications.
Transforming Chemical Analysis and Materials Discovery
The implications for industrial applications are substantial. Infrared spectroscopy is a workhorse technique across pharmaceuticals, materials science, and chemical manufacturing. Currently, interpreting complex IR spectra requires expert knowledge and often complementary techniques. A system that can accurately predict spectra from molecular structure could automate much of this analysis. More importantly, it enables inverse design—starting with desired spectral properties and working backward to molecular structures. This could accelerate discovery of new materials with specific optical properties, catalysts with optimized activity, or pharmaceuticals with targeted binding characteristics.
Technical Challenges and Scaling Limitations
Despite the impressive results, significant challenges remain before widespread adoption. The current validation focused on small organic molecules in gas phase—real-world applications often involve complex solvents, interfaces, and larger molecular systems. The mean absolute error metrics, while impressive, need validation across broader chemical spaces. Additionally, the requirement for 50 picosecond molecular dynamics trajectories, while efficient compared to traditional methods, still represents substantial computational cost for large systems. The framework’s performance on molecules significantly different from its training set—the extrapolation capability—remains a critical test for real-world utility.
Future Research Directions and Applications
The most exciting near-term applications likely lie in hybrid approaches where PALIRS handles routine predictions and flags uncertain cases for traditional computational methods. This could create a tiered system where computational resources are allocated efficiently based on prediction confidence. The methodology could also be extended to other spectroscopic techniques like Raman or NMR spectroscopy, creating a comprehensive computational spectroscopy platform. As the framework scales to larger molecules and more diverse chemical environments, it could become an essential tool in the computational chemist’s toolkit, much like density functional theory is today.
Broader Impact on Scientific Methodology
This research represents more than just another machine learning application—it signals a shift in how computational science approaches complex problems. The combination of active learning with ensemble methods and careful uncertainty quantification provides a blueprint for other scientific domains facing similar data-efficiency challenges. From protein folding to catalyst design, the principles demonstrated here could be adapted to numerous scientific challenges where traditional computational methods are prohibitively expensive. The success of PALIRS suggests we’re entering an era where machine learning doesn’t just accelerate existing methods but enables entirely new approaches to scientific discovery.